Our C++ code "SimSnappNet" is build on the software "SimSnapp" written by David Bryant and Remco Bouckaert.
D. Bryant, R. Bouckaert, J. Felsenstein, N.A. Rosenberg, A. RoyChoudhury : Inferring Species Trees Directly from Biallelic Genetic Markers: Bypassing gene trees in a full coalescent analysis. (Molecular Biology and Evolution, Volume 29, Issue 8, 2012)
download SimSnappNet from Github
SimSnappNet generates count data. The gene tree is random and is obtained according to the Multispecies Network Coalescent (i.e. a coalescent process that evolves inside a phylogenetic network). Next, SNPs data are obtained from a Markovian process evolving along gene tree branches. Finally, SNPs data are summed up in count data.
SimSnappNet requires a phylogenetic network in the
extended newick format ,
with branch lengths in substitution unit (see the remark at the bottom of this page).
Theta values (i.e. population sizes) are indicated in square brackets following node.
We used the following notation. The reticulation node is considered as a species, its name has to begin with #, followed by one single letter,
and the probability of going left. The sample size must be set to 0 for that node (see examples below!)
Optional insert
How to run SimSnappNet
./simsnap [-c] [nsites] [seed] filename
Flags are:
-c Include constant sites (default is to simulate only polymorphic sites, i.e. without -c it will generate polymorphic sites)
nsites: number of sites
seed: seed number for generating random data
filename: see below
Input File Format
number of species
species1-name sample-size
species2-name sample-size
...
species_n-name sample-size
mutation rate u mutation rate v
number of networks
network in extended newick format
Be careful, in your network in extended newick format, the fake leaf (referring to the reticulation node) has to be the second element in the cherry, e.g. (something,#H0.5) if we call the reticulation node #H0.5.
Besides, in your network in extended newick format, the internal reticulation node (above a node B), has to be in second place, e.g. (A,(B)#H0.5) and not ((B)#H0.5,A). In other words, the parent of A has two children, child 1 is A and child 2 is #H0.5.
Last, we have to mention that SimSnappNet can not handle loops, i.e. situations where the reticulation node has only one parent.
First example
Let us consider a network with 3 species, denoted A, B and C. Furthermore, on this example, 2 lineages belong to each species. Let us call #H0.5 the reticulation node of the following network. As previously said, we have to specify that the sample size associated to this fake species is equal to zero. 0.5 is the probability of going on the left side. On this example, the mutation rates u and v are both equal to 1.
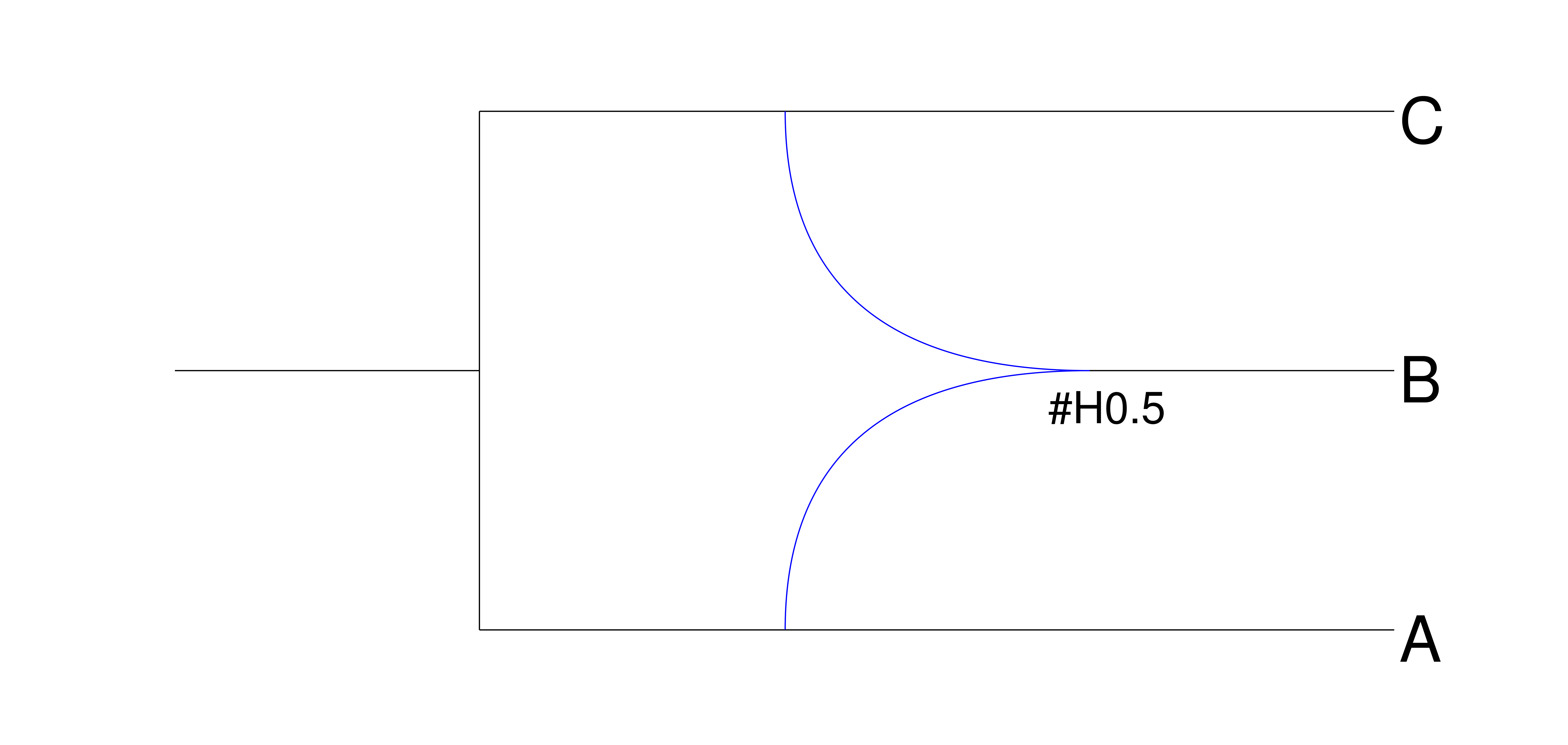
4
A 2
B 2
#H0.5 0
C 2
1.0 1.0
1
<1.0>((A[0.005]:0.035,(B[0.005]:0.012)#H0.5[0.005]:0.023)[0.005]:0.05,(C[0.005]:0.035,#H0.5[0.005]:0.023)[0.005]:0.05)[0.005]:0.1;
Download the associated file test_Net3species_A2B2C2
Second example (Network A)
Let us consider a network with 5 species, denoted C, R, Q, A and L. Furthermore, on this example, 2 lineages belong to each species. Let us call #H0.3 the reticulation node of the following network. As previously said, we have to specify that the sample size associated to this fake species is equal to zero. 0.3 is the probability of going on the left side. On this example, the mutation rates u and v are both equal to 1.

6
C 2
R 2
Q 2
A 2
#H0.3 0
L 2
1.0 1.0
1
<1.0>(C[0.005]:0.08,((R[0.005]:0.007,(Q[0.005]:0.004)#H0.3[0.005]:0.003)[0.005]:0.035,
((A[0.005]:0.006,#H0.3[0.005]:0.002)[0.005]:0.016,L[0.005]:0.022)[0.005]:0.02)[0.005]:0.038)[0.005]:0.1;
Download the associated file test_NetA_C2R2Q2A2L2
Third example (Network B)
An example with two reticulation nodes : #H0.3 and #E0.6
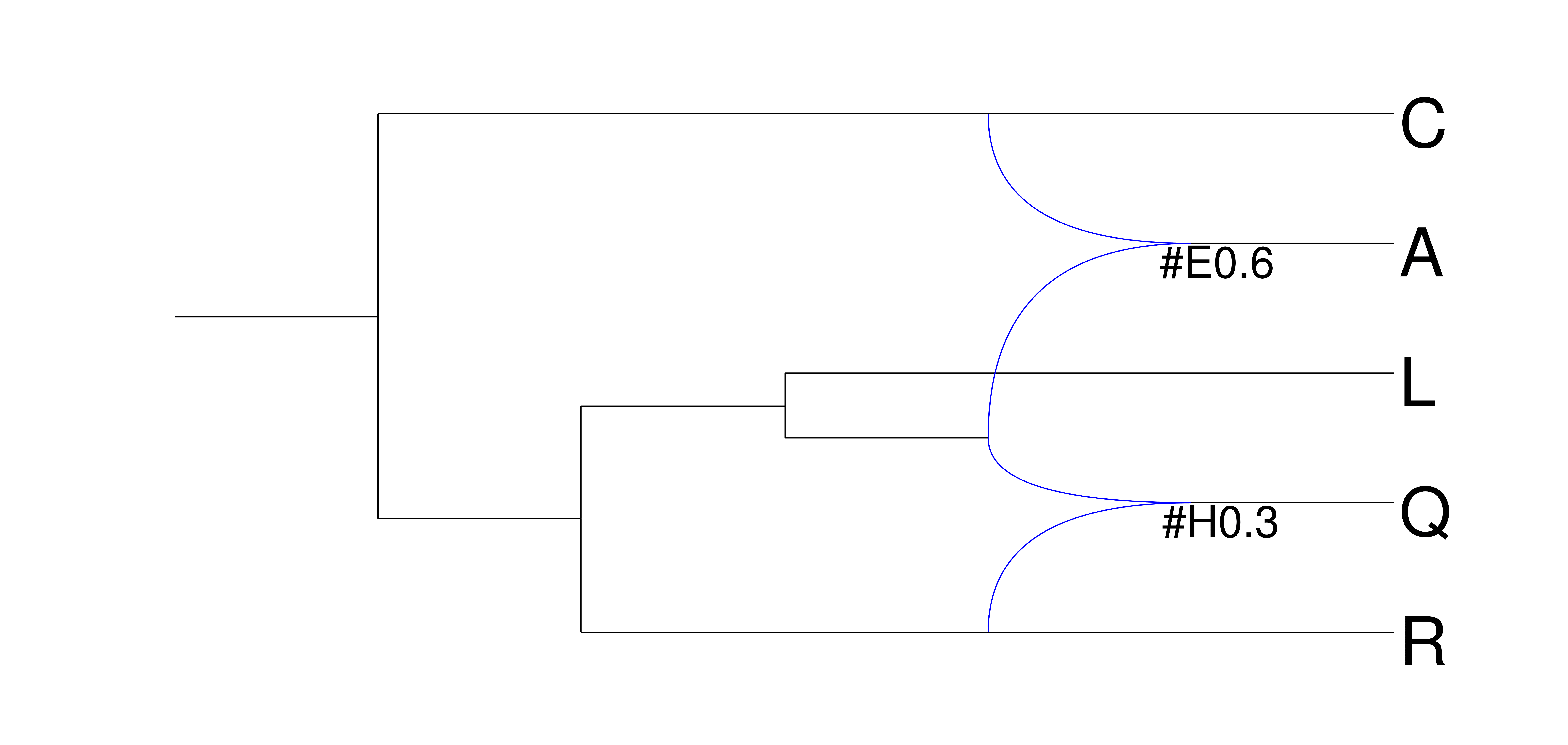
7
C 2
R 2
Q 2
A 2
#H0.3 0
L 2
#E0.6 0
1.0 1.0
1
<1.0>(((R[0.005]:0.014,(Q[0.005]:0.004)#H0.3[0.005]:0.01)[0.005]:0.028,(((A[0.005]:0.003)#E0.6[0.005]:0.003,#H0.3[0.005]:0.002)[0.005]:0.016,L[0.005]:0.022)[0.005]:0.02)[0.005]:0.038,(C[0.005]:0.005,#E0.6[0.005]:0.002)[0.005]:0.075)[0.005]:0.1;
Download the associated file test_NetB_C2R2Q2A2L2
Fourth example (Network C)
Another example with two reticulation nodes : #H0.5 and #E0.5
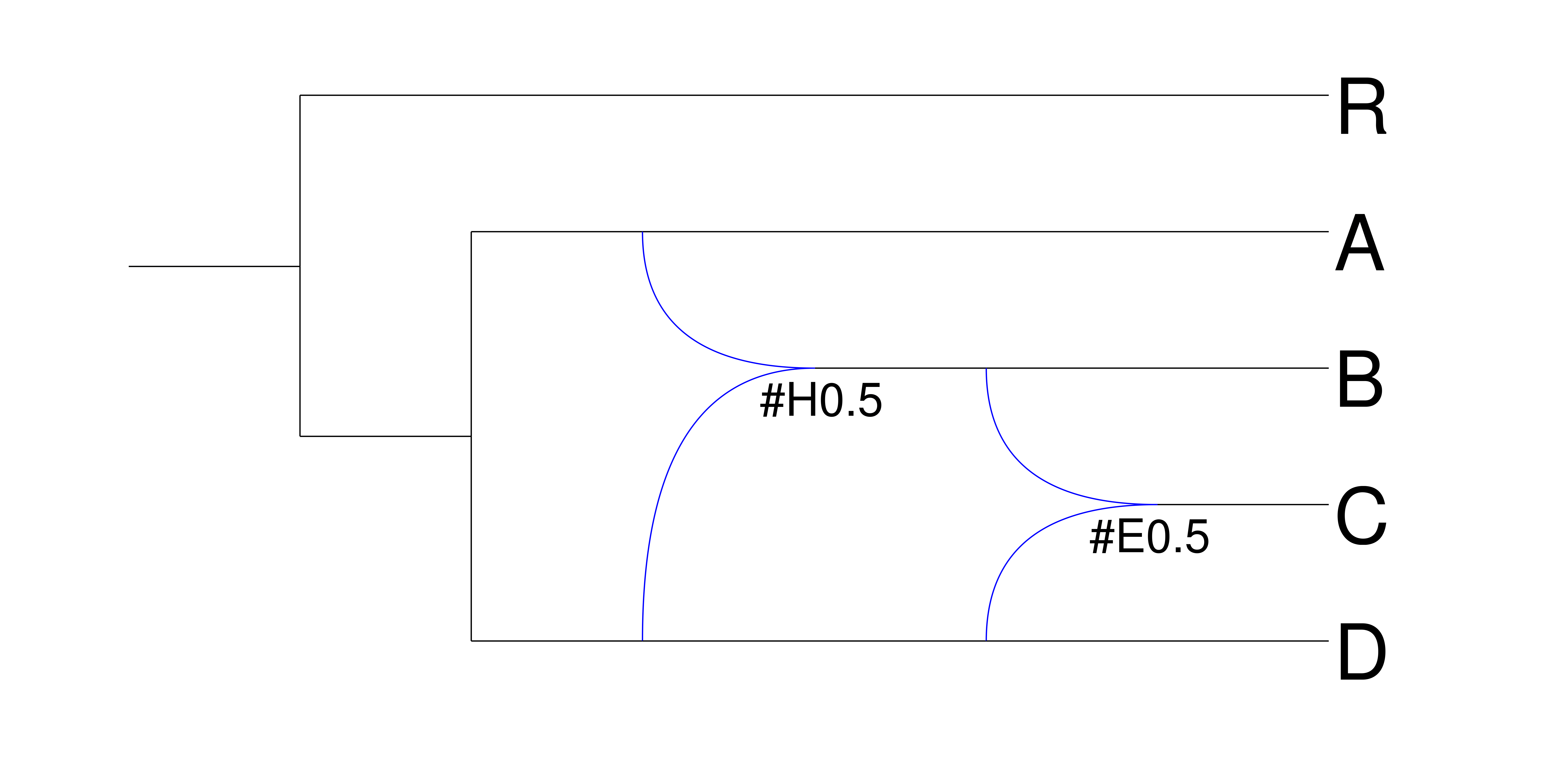
7
R 1
A 1
#E0.5 0
B 5
#H0.5 0
C 5
D 1
1.0 1.0
1
<1.0>(R[0.005]:0.08,((A[0.005]:0.012,((B[0.005]:0.002,(C[0.005]:0.001)#E0.5[0.005]:0.001)[0.005]:0.002)#H0.5[0.005]:0.008)[0.005]:0.038,((D[0.005]:0.003,#E0.5[0.005]:0.002)[0.005]:0.017,#H0.5[0.005]:0.016)[0.005]:0.03)[0.005]:0.03)[0.005]:0.1;
Download the associated file test_NetC_R1A1B5C5D1
Read the README file (available on github), to prepare the xml for Network C
Extra informations on the stochastic model
Gene tree model
For each site, the associated gene tree is obtained according to the Network Multispecies Colaescent Network model. The process starts at the leaves of the network and goes backward in time, until all lineages coalesce. At the beginning, coalescence occurs only between lineages that belong to the same species. 2 given lineages coalesce at rate 2mu/theta where theta and mu denote the population size parameter of that species , and a mutation rate, respectively. The parameter mu is equal to 2uv/(u+v) where u and v are mutation rates given in the input file. Assuming that k lineages belong to that species, the first coalescent time follows an exponential distribution E(k(k-1)mu/theta), since the coalescence of each combination of 2 lineages is equiprobable. When k=2, the expected coalescent time is theta/(2mu). theta is the average number of mutations, separating 2 individuals.Mutation model
As in SNAPP, we consider biallelic markers and the colors red and green represent the two alleles. Markers evolve along the gene tree branches, according to a continuous time Markov chain, where u and v denote respectively the instantaneous rates of mutating from red to green, and from green to red. Under this model, on a branch of length T, there are on average 2uvT/(u+v) mutations. Then, the mutation rate mu is equal to 2uv/(u+v). Imposing the constraint 2uv/(u+v)=1, enables to measure branch lengths in substitutions per site (i.e. genetic distance). If you do not impose this constraint, a branch of length T will represent on average T times mu mutations. Up to you !Webdesign service by Sarkis. Outsourcing by FreelanceWebmarket.